
A Google Colab notebook now hosts a complete plasmid engineering suite that runs Biopython, NumPy, and Matplotlib without needing a terminal interface. The tool loads plasmid records, calculates sequence statistics, maps restriction sites, and designs primers using standard Python libraries. It starts with a synthetic offline plasmid so the workflow runs reliably without external dependencies, while still supporting optional NCBI GenBank fetching and local file uploads.
Setup and data loading
The code installs Biopython if missing and imports the required scientific and plotting libraries. It defines a plasmid configuration, a colour palette for different features, and a multiple cloning site sequence. A fallback synthetic plasmid ensures the tutorial works even without an internet connection. Helper functions load records from NCBI or GenBank files and normalise annotated sequence features into metadata suitable for drawing.
try:
import Bio
except ImportError:
import subprocess, sys
subprocess.run([sys.executable, "-m", "pip", "install", "-q", "biopython"], check=True)
import math, io, textwrap, random
import numpy as np
import matplotlib
import matplotlib.pyplot as plt
from matplotlib.patches import Polygon
from Bio import Entrez, SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.SeqFeature import SeqFeature, FeatureLocation
from Bio.Restriction import RestrictionBatch, Analysis, CommOnly
try:
from Bio.SeqUtils import MeltingTemp as _mt
except Exception:
_mt = None
EMAIL = "yo*@*****le.com"
ACCESSION = "L09137"
USE_NCBI = False
Entrez.email = EMAIL
TYPE_COLORS = {
"CDS": "#d1495b", "gene": "#c1666b", "rep_origin": "#2e86ab",
"promoter": "#e3a008", "terminator": "#8d99ae", "misc_feature": "#6a994e",
"primer_bind": "#9d4edd", "protein_bind": "#457b9d", "source": "#adb5bd",
}
def _color(ftype): return TYPE_COLORS.get(ftype, "#6a994e")
MCS = "GAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTT"
def _synthetic_plasmid():
random.seed(19)
body = "".join(random.choice("ACGT") for _ in range(2686))
seq = body[:400] + MCS + body[400:]
rec = SeqRecord(Seq(seq), id="pDEMO", name="pDEMO",
description="Synthetic offline demo plasmid (SpliceCraft-Colab)")
rec.annotations["topology"] = "circular"
rec.annotations["molecule_type"] = "ds-DNA"
def feat(s, e, strand, ftype, label):
f = SeqFeature(FeatureLocation(s, e, strand=strand), type=ftype)
f.qualifiers["label"] = [label]; return f
rec.features += [
feat(400, 400 + len(MCS), 1, "misc_feature", "MCS"),
feat(700, 1561, 1, "CDS", "AmpR"),
feat(1700, 2260, -1, "rep_origin", "ori"),
feat(2350, 2686, 1, "CDS", "lacZ-alpha"),
]
rec.features[1].qualifiers["product"] = ["beta-lactamase (demo)"]
return rec
def load_record():
if USE_NCBI and "@" in EMAIL and not EMAIL.startswith("you@"):
try:
h = Entrez.efetch(db="nucleotide", id=ACCESSION,
rettype="gbwithparts", retmode="text")
rec = SeqIO.read(h, "genbank"); h.close()
print(f"✓ Fetched {ACCESSION} from NCBI: {rec.description}")
return rec
except Exception as e:
print(f" NCBI fetch failed ({e}); using synthetic demo plasmid.")
else:
print(" USE_NCBI is off (or EMAIL not set) — using synthetic demo plasmid.")
return _synthetic_plasmid()
def upload_gb():
from google.colab import files
up = files.upload()
name = next(iter(up))
return SeqIO.read(io.StringIO(up[name].decode()), "genbank")
def label_of(f):
for k in ("label", "gene", "product", "note"):
if k in f.qualifiers: return f.qualifiers[k][0]
return f.type
def norm_features(rec, skip_source=True, min_len=0):
L = len(rec.seq); out = []
for f in rec.features:
if skip_source and f.type in ("source",): continue
s = int(f.location.start); e = int(f.location.end)
if (e - s) < min_len: continue
out.append(dict(start=s % L, end=e % L, strand=f.location.strand or 1,
type=f.type, label=label_of(f), color=_color(f.type)))
return out
NCBI fetch failed ({e}); using synthetic demo plasmid.")
else:
print(" USE_NCBI is off (or EMAIL not set) — using synthetic demo plasmid.")
return _synthetic_plasmid()
def upload_gb():
from google.colab import files
up = files.upload()
name = next(iter(up))
return SeqIO.read(io.StringIO(up[name].decode()), "genbank")
def label_of(f):
for k in ("label", "gene", "product", "note"):
if k in f.qualifiers: return f.qualifiers[k][0]
return f.type
def norm_features(rec, skip_source=True, min_len=0):
L = len(rec.seq); out = []
for f in rec.features:
if skip_source and f.type in ("source",): continue
s = int(f.location.start); e = int(f.location.end)
if (e - s) < min_len: continue
out.append(dict(start=s % L, end=e % L, strand=f.location.strand or 1,
type=f.type, label=label_of(f), color=_color(f.type)))
return out
 NCBI fetch failed ({e}); using synthetic demo plasmid.")
else:
print("
NCBI fetch failed ({e}); using synthetic demo plasmid.")
else:
print(" USE_NCBI is off (or EMAIL not set) — using synthetic demo plasmid.")
return _synthetic_plasmid()
def upload_gb():
from google.colab import files
up = files.upload()
name = next(iter(up))
return SeqIO.read(io.StringIO(up[name].decode()), "genbank")
def label_of(f):
for k in ("label", "gene", "product", "note"):
if k in f.qualifiers: return f.qualifiers[k][0]
return f.type
def norm_features(rec, skip_source=True, min_len=0):
L = len(rec.seq); out = []
for f in rec.features:
if skip_source and f.type in ("source",): continue
s = int(f.location.start); e = int(f.location.end)
if (e - s) < min_len: continue
out.append(dict(start=s % L, end=e % L, strand=f.location.strand or 1,
type=f.type, label=label_of(f), color=_color(f.type)))
return out
USE_NCBI is off (or EMAIL not set) — using synthetic demo plasmid.")
return _synthetic_plasmid()
def upload_gb():
from google.colab import files
up = files.upload()
name = next(iter(up))
return SeqIO.read(io.StringIO(up[name].decode()), "genbank")
def label_of(f):
for k in ("label", "gene", "product", "note"):
if k in f.qualifiers: return f.qualifiers[k][0]
return f.type
def norm_features(rec, skip_source=True, min_len=0):
L = len(rec.seq); out = []
for f in rec.features:
if skip_source and f.type in ("source",): continue
s = int(f.location.start); e = int(f.location.end)
if (e - s) < min_len: continue
out.append(dict(start=s % L, end=e % L, strand=f.location.strand or 1,
type=f.type, label=label_of(f), color=_color(f.type)))
return out
The script sets up the environment, installs Biopython when needed, and imports the scientific, plotting, and sequence-analysis libraries required for the workflow. It defines the plasmid configuration, feature colour palette, multiple cloning site, and fallback synthetic plasmid so the tutorial works even without an internet connection. Helper functions load records from NCBI or GenBank files and normalise annotated sequence features into drawable metadata.
Visualising circular maps
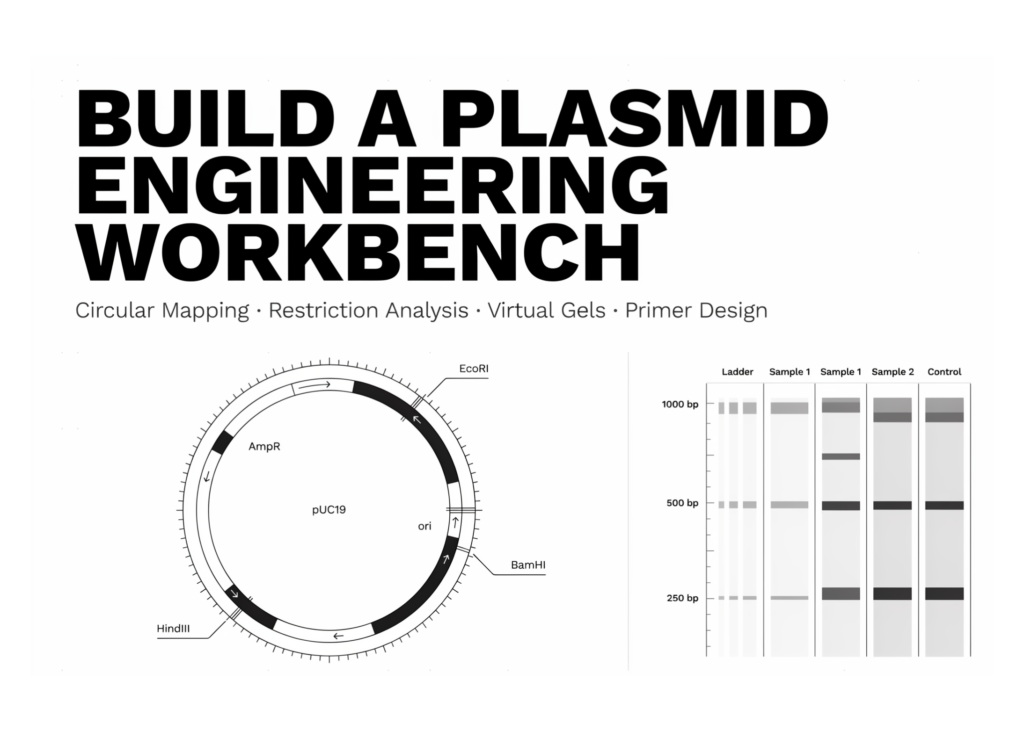
The circular plasmid visualisation maps base-pair positions to angular coordinates on a clockwise ring. The code calculates GC content, draws the plasmid backbone, adds tick marks, renders feature arcs, and attaches directional arrowheads to clearly indicate strand orientation. Labels and central sequence metadata appear so the map is both biologically informative and visually readable in Colab.
def _ang(bp, L): return math.pi/2 - 2*math.pi*(bp/L)
def _pt(bp, r, L):
a = _ang(bp, L); return r*math.cos(a), r*math.sin(a)
def _arc(s, e, r, L, n=240):
if e < s: e += L
bps = np.linspace(s, e, max(2, int(n*(e-s)/L)+2))
return ([r*math.cos(_ang(b, L)) for b in bps],
[r*math.sin(_ang(b, L)) for b in bps])
def gc_percent(seq):
s = str(seq).upper(); n = len(s) or 1
return 100.0 * (s.count("G") + s.count("C")) / n
def circular_map(rec, title=None, show_gc=True):
L = len(rec.seq); feats = norm_features(rec)
fig, ax = plt.subplots(figsize=(8, 8)); R = 1.0
if show_gc:
w = max(30, L // 120); step = max(1, w // 2); mean = gc_percent(rec.seq)
s = str(rec.seq).upper()
for i in range(0, L, step):
win = s[i:i+w] or s[i:] + s[:(i+w) % L]
dev = (gc_percent(win) - mean) / 100.0
rr = 0.72 + dev * 0.9
x0, y0 = _pt(i, 0.72, L); x1, y1 = _pt(i, rr, L)
ax.plot([x0, x1], [y0, y1],
color="#2e86ab" if dev >= 0 else "#d1495b", lw=1, alpha=0.5, zorder=1)
xs, ys = _arc(0, L, R, L); ax.plot(xs, ys, color="#333", lw=2.5, zorder=2)
step = max(1, round(L/12/100)*100) or max(1, L//12)
for t in range(0, L, step):
x0, y0 = _pt(t, R*1.015, L); x1, y1 = _pt(t, R*1.045, L)
ax.plot([x0, x1], [y0, y1], colorSource Read original →Related articles

